病因
病因
病因:胸
腹裂孔疝的发生与腰肋三角部发育延缓或融合不全有关,但其发育延缓或融合不全的原因不明,目前认为是由遗传因素和环境因素的相互作用所致。胸
腹裂孔疝的疝环为膈肌发育延缓或融合不全所形成膈肌缺损,大多位于左后外侧,其缺损裂口呈三角形、卵圆形或裂隙形,裂隙的尖端指向中心腱,底边在胸侧壁肋缘处,边缘光滑、增厚,触之有光滑的棱状感。缺损范围(疝环)大小不一,小者仅有1cm,大者可占整个半侧膈肌,甚至一侧膈肌几乎完全缺如,只有胸肋部一些残存膈组织。多数病例缺损前侧的膈肌发育尚好而后侧发育不良,或因膈肌缺损过大后侧完全缺失。缺损过大者手术修补膈肌非常困难,术后亦容易复发。胸
腹裂孔缺损(疝环)大者一般不发生嵌顿,疝环较小者则易发生嵌顿,致使进入胸腔内的肠管膨胀扩大,压迫肺和心脏,影响呼吸和循环。
胸
腹裂孔疝10%~15%的病例有疝囊,无疝囊者占绝大多数(85%~90%)。如在胚胎8周前胸
腹裂孔尚未闭合前腹腔脏器已滑入胸腔者,或胸
腹裂孔闭合延迟甚至未闭合者即无疝囊。膈肌的形成只有胸膜和腹膜的胸腹膜皱褶而无肌层,腹腔脏器突向胸腔者即有疝囊。疝囊由胸、腹膜两层浆膜构成,中间可见一些结缔组织或稀少的肌肉组织,呈薄膜状。有疝囊的胸
腹裂孔疝,由于受疝囊的限制,进入胸腔的腹腔脏器多寡、体积相对较少,因而胸腔内脏器受影响的程度也较轻;相反,无疝囊者,大量肠管等腹腔脏器可进入患侧胸膜腔,胸腔内脏器受影响的程度也较重。此外,疝囊的有无亦与肺发育受影响的程度直接有关,有疝囊者受影响相对较小,预后较好;反之,则影响大,预后差。
左侧胸
腹裂孔疝的内容物最常见的是小肠,其次是胃,其它还有结肠和脾脏等;由于膈肌的发育和中肠的发育及旋转几乎同时发生,如在胚胎8周前胸
腹裂孔尚未闭合前腹腔脏器已滑入胸腔,或在胚胎10~12周时因胸
腹裂孔闭合延迟而使中肠退回腹腔同时进入胸腔,可导致中肠旋转不良,盲肠与阑尾则进入胸腔;部分病人的疝内容物仅为胃、脾和肝脏的一部分,少数病人还可有腹膜后的肾脏、肾上腺进入胸腔内。右侧胸
腹裂孔疝,肝脏往往是疝入胸腔的惟一脏器,少数有结肠和小肠。与创伤性膈疝不同的是,
先天性膈疝的疝内容物很少与胸腔内器官发生
肺发育不良也是
先天性膈疝的重要病理改变之一。肺的正常发育开始于胚胎第4周,根据组织形态所见肺发育可分4期:胚胎期,从胚胎第4周开始到第5周,此期气管末端首先分叉形成两侧肺芽;假腺体期,胚胎第5~17周,肺芽不断分支形成支气管树。肺动脉及其分支也伴行各级支气管分支发育,支气管树的连续分支在胚胎第16周基本完成。在这一过程中,各级支气管尚未向外界开放,内衬连续的上皮,周围为实质性间质,故胚胎学家称该期为假腺体期;小管期,胚胎第16~24周,毛细血管向支气管树终末盲端周围的上皮细胞间长入,使盲端的上皮被间断,发育成呼吸性细支气管和囊泡,支气管树开始成为小管状,称为肺发育的小管期;终末囊泡或肺泡期,胚胎第24周至出生后一段时期。胚胎第16周后非呼吸性支气管分支不再增加,而肺泡则开始并继续分化发育直至生后8岁左右,其数量出生时约2000万,出生后肺泡数量继续增加,至8岁其总数可达出生时的10倍。支气管树分支完成后其管道逐渐延长,管径增大,肺芽周围的间质分化成各级支气管壁中的软骨、平滑肌等组织。
如胚胎第8~9周膈肌未闭合,肠襻进入胸腔,压迫正在发育分支的支气管和肺动脉,则阻碍其正常发育,所导致的病理改变与膈疝疝环的大小有关。尸检出生后因膈疝死亡的新生儿发现,双肺均发育障碍,肺支气管分支数少,肺泡数量亦明显减少,外观呈萎缩状,体积小、重量轻。部分病儿肺发育不全和
肺不张同时存在,有时对侧肺亦可有不同程度的发育不良。患侧与10~12周胎龄的胚胎肺相当,而对侧则仅与12~14周胎龄的胚胎肺相仿。患侧肺段自肺门至远端胸膜的支气管的分支数仅为正常新生儿的一半,而对侧较正常的一半略多一点,肺泡数量仅为正常新生儿的1/3。由于肺动脉分支的发育伴随支气管分支同时进行,因此两者发育可同受阻碍。肺动脉的数量、分支明显减少且较细长,肌层增厚;发育不全的左下肺体积及其动脉的管径均处于胎儿型,与胚胎第12~14周时的情况相当;囊泡前小动脉壁中的平滑肌层较正常刚出生的新生儿肥厚,且患侧的肺小动脉的肌层又较对侧肥厚。
此外,一些学者通过动物及人体实验发现,本病患儿肺泡表面活性物质较正常新生儿少,可导致肺泡张力增高、肺顺应性降低和通气能力下降。但亦有人对患儿是否缺少肺泡表面活性物质有异议。
有些先天性胸腹裂孔疝的病儿可伴有其他先天性畸形,如
先天性肠旋转不良和先天性心脏病等。Bollmann等通过对33例经产前诊断为先天性胸腹裂孔疝患儿的观察,发现其中24例同时合并1种或2种以上畸形,包括心血管系统、运动系统、泌尿生殖系统及神经系统畸形;其中,6例合并
染色体异常,如18三体综合征。
发病机制
发病机制:由于胚胎期胃、肠管等大量腹腔脏器进入胸腔,肺脏受压,发育受阻(肺支气管分支数和肺泡数量大大减少),新生儿出生后呼吸功能欠佳,表现为呼吸困难。生后开始呼吸,随着吞咽的空气进入胃肠道,进一步加重了对肺的压迫,阻碍气体交换,出现动脉氧分压降低、二氧化碳分压升高、
呼吸性酸中毒。缺氧、酸中毒可引起
肺血管痉挛,导致肺动脉阻力增高,血液经动脉导管和卵圆孔由右至左的分流量增加,进一步加重低氧血症和酸血症,并使之形成恶性循环。
胎儿期,右心室排出的血液有85%未通过肺,而直接经卵圆孔或动脉导管分流入主动脉至胎盘进行气体交换。胎儿型循环的分流是由于胚胎肺内充满液体和肺小动脉因血氧低而收缩使肺血管阻力增高所导致的。新生儿出生后肺内充满空气、肺泡内液体被吸收、肺小动脉因氧分压增高而扩张致使肺血管阻力随即降低,加之断脐后体循环血压升高,循环即转为成人型循环。出生后有3种肺血管发育异常可引起肺血管阻力增高并发生肺外右至左分流,其一:肺血管广泛细小,使肺血管的总血容量减少;其二:肺动脉分支减少,使单位肺组织内的血管数减少;其三:肺小动脉的平滑肌层增厚。
由于
先天性膈疝伴发肺小动脉发育不良,管壁肌层肥厚,对低氧和高碳酸血症非常敏感,更易发生血管痉挛,这是患儿
肺动脉高血压和右至左分流的主要原因。除了
肺血管痉挛因素外,肺脏发育不良、肺血管普遍细小、肺动脉分支数量少等因素直接导致肺血管内总血容量减少,也是引起肺血管阻力增高、发生血循环右至左分流的重要原因。
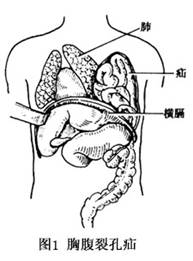
疝环(膈肌缺损)过大者极少发生嵌顿,小者则易发生嵌顿。当咳嗽、哭闹或用力排便时可使腹内压升高,致使较多胃肠管进入胸腔内。由于疝环的卡压,肠管排空障碍,严重者发生绞窄。随着肠壁水肿和肠腔膨胀扩大、对肺和心脏的压迫加重,呼吸和循环逐渐发生障碍。如果嵌顿的肠管发生血循环障碍,出现绞窄坏死、穿孔时,可导致严重的胸腹腔感染、中毒性休克(图1)。
其他辅助检查
其他辅助检查:
1.X线检查
(1)X线透视:胸腔内可见疝入肠襻及其蠕动影像。
(2)X线摄片:消化道造影及X线摄片检查对本病的诊断起决定性作用。摄片应包括胸腹部,其X线平片影像有:①患侧正常膈肌的弓形影像不清、中断或消失;②患侧肺萎陷;③纵隔、心脏向健侧移位;④胸腔可见疝入腹腔脏器影像:A.疝内容物单纯为胃时,腹部见不到腹腔内的胃泡影像,而胸腔内则显示有较大的含液气面的胃泡;B.疝入胸腔的内容物为小肠且小肠未充气时,胸腹部X线平片可见患侧胸腔内有密度增高阴影,随着病儿开始呼吸、吞咽的空气进入肠道后,反复检查胸腔内可显示有扩张积气的肠襻影。如小肠充气膨胀后其X线平片可见有积气、透亮的肠襻,而且其影像向腹部延续;C.疝内容物为肝脾等实质性脏器时,除在胸腔内见到实质性影像外,可见肝影上移及上移后留下的空隙有肠管充填。
(3)消化道造影:高度怀疑胸腹裂孔疝,行胸腹部X线平片不能确诊者,可选择消化道造影。
方法:首先经鼻置入胃管,然后向胃管内注入少许空气或水溶性造影剂(如12.5%的碘化钠),如发现胸腔内有胃肠道影像,即可做出诊断。
新生儿和婴幼儿以选择空气或水溶性造影剂为宜,禁用钡剂造影,以防误吸导致窒息死亡,年长儿可选用钡剂造影。
(4)CT检查:可比较清晰的显示疝环(膈肌缺损)的边缘和疝入胸腔内的腹腔脏器影像,疝环横断面呈三角形,其内影多呈肠管蜂窝状。
2.B超检查 可发现胸腔内有扩张的肠襻以及其蠕动回声,或伴有液体无回声及气体点状回声的游动影,肠段积液时可见粘膜皱襞。
近年来,国内外学者依靠超声检查开展了本病的产前诊断研究。产前超声检查时如能证实腹腔脏器位于胸腔内则可确定诊断,如发现羊水过多、纵隔移位、腹腔内缺少胃泡等征象时,应进一步详细检查是否有腹腔脏器疝入胸腔。
3.MRI检查 冠状面可清晰的显示疝入胸腔内肠管的影像及疝环的边缘。疝环横断面与CT影像相似,呈三角形,内有断面呈蜂窝状肠管影。
诊断
诊断:
1.临床特点 由于膈肌缺损大小、有无疝囊、腹腔脏器疝入胸腔的多寡、肺受压萎缩发育不良程度及健侧肺发育障碍的状况不一,临床症状出现的早晚有很大的差异。
(1)症状:新生儿生后即出现临床症状者,往往膈肌缺损大、无疝囊、疝入胸腔的腹腔脏器多、患侧肺受压萎缩发育不良及健侧肺发育受影响的程度较重,症状重、变化快、病情凶险,甚至出生后短期之内因未能及时确诊和治疗而死亡。对此,早期诊断、及时治疗,是减少死亡的关键。如新生儿出现急性呼吸困难、短促和青紫,喂奶、哭闹后加重,应高度怀疑本病。婴幼儿和儿童期临床症状与体征不尽相同,对反复发生呼吸道感染或反复出现咳嗽、气促,或随体位变动、哭闹、饱食和活动之后出现呼吸急促、呼吸困难和发绀、
腹痛和呕吐者,亦须考虑本病。
(2)体征:呼吸急促且费力,发绀;患侧胸廓饱满、肋间隙增宽;呼吸动度变弱;叩诊浊音鼓音相间;听诊患侧肺下野呼吸音减弱或消失并可闻及肠鸣音。
2.影像学检查 除症状和体征外,先天性胸腹裂孔疝的X线影像有以下特点:
(1)X线透视下胸腔内可见疝入的肠襻及其蠕动影像。
(2)胸腹部或胸廓及上腹部X线平片显示:膈肌弓形边缘的影像中断、不清或消失,胸腔内含有液气面或积气肠管蜂窝状影像、且这种影像胸腹腔相连续,患侧肺萎陷,心脏和纵隔向健侧移位。
(3)消化道造影示胸腔内有胃肠道影像。
(4)CT和MRI扫描可显示疝环的边缘及其呈三角形的横断面内有蜂窝状肠管影像。
(5)B超可发现胸腔内有扩张的肠襻以及其蠕动回声。
鉴别诊断
鉴别诊断:
1.先天性肺囊肿 ①本病是胚胎初期肺芽由实心索状向中空成管状发育的过程发生障碍,近端阻塞,远端原始支气管形成盲囊,囊内的分泌黏液不能排出,逐渐扩张形成以支气管组织为壁的囊肿。有单发性和多发性肺囊肿之分。与支气管不相通者为闭合囊肿,囊腔被黏液充盈。与支气管相通者为开放囊肿,黏液经细小通道排出气管,支气管与囊腔间有时形成一个单向“活瓣”,吸气时空气较易进入囊腔并使其膨胀,呼气时囊内气体很难排出而成为张力性囊肿,压迫患侧正常肺组织而且使纵隔及心脏移位、对侧肺亦受压,呼吸功能受损;②本病多在青、壮年期发病,新生儿期发病者少见;③新生儿期出现症状者多为开放囊肿,初期表现呼吸急促,随之呼吸呈吸气性呼吸困难状并持续加重,缺氧发绀、烦躁不安、头部出冷汗,伴发感染后患儿有多痰、发热,但无消化道症状;④有明显的“三凹”征和胸内压增高征象。但肝或脾浊音界下移,腹部不凹陷,胸部听诊无肠鸣音;⑤X线胸片上可见明显的“胸内积气”征,患侧膈肌弓形影像完整,囊壁薄,囊肿影像不向腹腔侧延续,消化道造影胸腔内无胃肠道影像。
2.有疝囊的胸
腹裂孔疝与膈膨升的鉴别 ①膈膨升有先天性膈膨升和后天性膈膨升之分,前者为胚胎期膈肌发育障碍,一侧或双侧、部分或完全性薄弱所致;后天性膈膨升是因外伤(如产伤损伤颈3、4、5神经根),膈神经
麻痹而致;②极少合并肺发育障碍;③与先天性胸腹裂孔疝的症状相似,但后天性膈膨升有难产或产伤史,多系臀位产,多数患儿同时合并Erb氏
麻痹、
锁骨骨折、肱骨骨折等;④胸腹部X线片特点为,膈肌阴影明显升高,膈顶呈弓形,膈肌呈完整而光滑的弧形阴影,膈下方有胃泡或肠管影或肝脏阴影。但局限性膈膨升和胸
腹裂孔疝很难鉴别,只要症状明显、有手术指征,可手术确诊并治疗。
3.其它 尚需与肺炎所致的肺大泡、脓
气胸相鉴别,只要详细询问病史、仔细查体,结合影像学检查一般不难鉴别。
治疗
治疗:确诊后首选尽早手术修补治疗,以免日后形成粘连或并发绞窄性疝。绝不能依赖药物和内科处理。症状较轻或病情稳定的较大儿童,可适当放宽,选择择期手术。
术前准备包括:①插胃管行胃肠减压,防止麻醉和手术过程中胃肠道含气量增多疝入胸腔的脏器体积更加增大或发生气胸,影响术野,甚至会加重呼吸循环功能障碍。②新生儿行脐动脉插管,以利血气监测、输液和用药。③病情危重的新生儿应尽快气管插管并行机械呼吸[氧吸入浓度100%,气道压<3.3kPa(25mmHg),PEEP 0.490kPa(5cmH
2O)。若有严重的呼吸性酸中毒,则行过度通气(100~150/min)并输入碳酸氢钠。④长时间的低血氧症、酸中毒、碳酸过多症会导致肺血管痉挛及持续胎儿循环状态。若心脏功能不好,可用
多巴酚丁胺(dobutamine)支持。为改善肾脏的灌注,可用小量的多巴胺(dopamine)。⑤如有低血压,则输入
人血白蛋白(
白蛋白)。
手术究竟采用胸部或腹部切口,意见尚不完全一致。多采用经腹途径,取左上横切口或旁正中切口。开腹后将疝入胸腔的脏器轻柔地拉回腹腔。若缺损小,可适当剪开扩大,以利脏器回纳。通过缺损,检查肺的发育情况,注意不可过度加压呼吸促使其膨胀。缺损可用间断缝合法修补,若缺损后缘缺如,则应将缝线缝在缺损后缘相邻的肋骨上,以保证修补牢固、可靠。缺损过大而不能直接缝合对拢时,可用人工合成材料(如marlex、polytetrafluoroethylene膜)修补。腹腔内合并的其他畸形,如肠道转位不全、腹膜纤维带压迫所致的幽门梗阻和十二指肠梗阻等,术中应一并矫正。若腹腔容积小,容纳不下还纳的腹腔脏器,或勉强缝合腹壁切口后影响呼吸时,则用硅橡胶片建造临时性人工腹壁疝。术后是否在第8或第9肋间腋中线安置胸腔引流管仍有争议。安置引流管有利于胸内气体和液体的排出。
较大的儿童,或术前已明确胸内有病变需要矫治者(如合并肺外型隔离肺),或估计胸内有广泛粘连者,则采用侧卧位经胸途径手术。
术后处理:①继续机械呼吸,过度通气,使之成为呼吸性碱中毒状态。当生命体征突然恶化时,应注意健侧胸腔有无张力气胸发生。若有,则应立即行胸腔闭式引流。②保持胃肠减压通畅。③动脉血气监测常显示术前的代谢性酸中毒仍未纠正,可继续输入碳酸氢钠溶液。④严格控制输液总量。⑤肺发育不全严重的婴幼儿,术后肺循环仍会维持在胎儿状态,肺动脉压很高,经未闭动脉导管产生的右向左分流仍然存在。这类婴幼儿术后12~36h内情况明显好转,但不久又恶化。此时可应用肺血管扩张药
妥拉唑啉(tolazoline)、一氧化氮及心肌收缩增强药。患儿能否存活,将取决于肺动脉高压改善的情况及肺功能的好坏。近十几年,膜式肺呼吸支持挽救这些重症婴幼儿的报告陆续出现,可以探索、尝试。
预后
预后:先天性胸腹裂孔疝的预后取决于膈肌缺损程度、疝入腹腔脏器的多寡、肺发育不良的程度、疝入胸腔的脏器对肺和纵隔压迫程度、
肺动脉高压及由左至右分流的程度、治疗是否及时和恰当等多种因素。如膈肌缺损大、疝入的腹腔脏器多,胚胎期对患侧肺和对侧肺发育的影响就大,出生后即可发病,预后较差;反之,肺发育受影响的程度就小,多在婴儿或幼儿期发病,预后相对较好。
1.发病时间与病死率 临床研究证实,出生后能够正常呼吸超过6h的患儿,其肺发育障碍的程度多不严重。而出生6h内发病的危重病儿为高危型,其病死率非常高。伍连康报道,症状在生后6h内出现者,病死率为54.5%;在生后6h后出现症状者,病死率为18.2%。甚至有的学者报道,出生6h内发病的危重病儿的病死率高达73%。出生时稍有或完全没有呼吸困难,直至婴儿期或儿童期后才发现有胸
腹裂孔疝存在者,其成活率非常高。
2.肺发育与预后 大量临床研究发现,出生时表现正常、无显著肺发育不全和呼吸困难,只在胸内胃肠充气压迫严重时才出现者,术后存活率75%~90%。严重双肺发育不全,出生数小时内停止呼吸,偶有存活时间超过6h者。pH<7.0、PaC02>60mmHg、Pa02<50mmHg的病例,病死率极高。
一些病儿经过手术将进入胸腔的腹腔脏器复位并修补缺损,解除了腹腔脏器对肺脏的压迫,使压缩肺叶膨胀,呼吸可暂时获得明显好转,随后却因肺组织发育不良、顺应性差,呼吸功能依然欠佳,以及肺血流量少不足以进行有效的氧合作用,最终死于仍然严重缺氧和
呼吸性酸中毒。有学者报道1例生后即呼吸困难和发绀的左侧膈疝病儿,25h后行手术腹腔脏器复位并修补缺损、术后10周死亡。尸检发现:患侧肺体积仍小于同龄新生儿的肺脏,双肺肺泡内气体占肺体积的百分比大于正常比例,对侧肺底段终末细支气管的分支数仅为正常的2/3;双肺肺泡总数虽较出生时多,已发育的肺囊泡也按肺泡的正常发育速度增长数量,但因支气管分支减少而缺失的肺囊泡及其肺泡数确无法得到改善或增加。
对手术后存活者随访发现,手术后2周内患侧肺缓慢膨胀,8个月~2年后患侧肺虽扩张,但透光度高;20年后,肺功能测定和肺扫描检查发现,肺功能和肺血灌注量仍不足。表明:①先天性胸腹裂孔疝手术后,压迫、限制肺发育的因素虽已消除,胚胎期肺发育的程度仍然是影响预后的主要因素。②出生后手术解除对肺脏的压迫后,肺泡过度充气所引起的肺体积的突然增大,对气体交换并非有利。③支气管的分支数不能增加,肺泡总数也不能增加。④发育不全的肺过度充气,由于肺的灌注不足,不能增加气体的交换。
3.
肺动脉高压与预后 先天性胸腹裂孔疝所伴有的肺小动脉异常,特别是管壁肌层肥厚,对酸血症、低氧和高碳酸血症非常敏感易引起痉挛,导致肺血管阻力增高并引起右至左分流,进一步加重低氧和高碳酸血症,并形成恶性循环,严重威胁病儿生命。手术解除肺的压迫后可使其氧合作用得到一定的改善,但肺动脉分支减少、肺小动脉的平滑肌层增厚的病理改变仍存在,由此引起的
肺血管痉挛导致的右至左分流、缺氧亦存在。
近年来,临床上应用血管扩张药物治疗
肺动脉高压,可提高本病的存活率。但是,肺实质和肺血管发育严重不全的重危病儿,其肺血管及其分支数量显著减少和其管腔细小,产生持续性低血氧继发
持续胎儿循环,仅解除肺小动脉痉挛并不能改善氧合作用。只有肺血管发育不严重者,肺小动脉痉挛、肺循环阻力增高引起的
肺动脉高压和右至左分流是影响预后的关键性的因素时,应用扩张血管药物才能改善氧合作用、提高存活率。
波士顿儿童医院对高危病儿手术后常规行心导管和肺血管造影,以了解肺血管的发育和血液动力学确切变化。他们将:①严重的持续性主动脉导管后低血氧、肺血管床不能容纳正常的心搏出量、各种治疗都无法增加肺灌注量、大部分右心排出血量分流入降主动脉者,称为无反应型。②术后出现“蜜月期”、肺血管床有容纳全部甚至更多的心搏出血量的潜力、肺血管阻力正常、术后出现轻度
肺动脉高压、但能保持正常氧合作用者称之为反应型。前者患儿的动脉缺氧多进行性加重且全部死亡。后者病儿术后亦可常因多种因素刺激诱发严重
肺动脉高压而导致右至左分流,如不进行及时有效的处理亦可导致死亡。在治疗过程中大多数反应型患儿在术后可多次发生肺动脉压暂时高于主动脉压,出现经动脉导管的右至左分流,但经调节呼吸机和加深麻醉后都恢复正常,随着肺动脉压下降,动脉导管常于出生后10~30h自行闭合。
4.产前诊断与预后 在胎儿期超声检查不仅能发现腹腔脏器位于胸腔内、确定胎儿胸
腹裂孔疝的诊断,而且能发现胸腔疝入脏器的多寡,可作为判断预后的重要手段和指标。Bronshitein等对15例经产前诊断为
先天性膈疝病儿的观察发现,产前诊断时间与预后相关,诊断时间大于25周的胸
腹裂孔疝患儿预后良好;产前诊断时间越早,预后越差。
5.产前治疗与预后
(1)产前激素治疗与预后:Losty等对猫
先天性膈疝模型产前试用激素治疗,包括地塞米松、促甲状腺释放激素、地塞米松-促甲状腺释放激素治疗,以生理盐水为对照,监测指标包括总内表面积、气腔容积比、气道比例等。并发现地塞米松、地塞米松-促甲状腺释放激素治疗组各项肺成熟度的形态学监测指标较对照组均有明显改善,预后也较好。但对产前诊断为
先天性膈疝者,是否使用激素治疗尚未见报道。
(2)宫内手术治疗与预后:动物实验证实,在妊娠期间手术将进入胸腔的腹腔脏器回复并修补膈肌缺损,肺的发育足以在出生后支持生命。胎儿通过开放性手术回复腹腔脏器、修补膈肌缺损的治疗尚处在实验阶段,而且技术要求非常高。Harrison等人曾手术治疗14例在妊娠24周前作出诊断、未合并其它畸形的严重膈疝胎儿,5例在手术期间死于技术原因,9例手术成功的胎儿中4例出生后存活,3例术后48h内死于宫内,2例
早产死亡。相信,随着医疗技术水平的提高,产前诊断和宫内手术将成为降低病死率、改善肺发育、提高患儿生后生活质量的非常有希望的一条途径。
6.围手术期处理与预后 过去主张,一些
先天性膈疝患儿在就诊时病情危重,尤其生后6h以内出现呼吸窘迫的高危患儿,应急症手术治疗。但经过大量临床研究发现,急症手术修补膈疝和内脏复位并不像预计的那样可改善肺功能,相反术后气道阻力的增加和肺顺应性的降低可使肺功能恶化和加重
肺动脉高压;而术前进行稳定治疗后,可以提高存活率和改善预后。因而近年来主张选择性地限期手术,病情危重者应采用体外膜式氧合、一氧化氮吸入和应用前列腺素降低持续性
肺动脉高压,待病儿术前心肺功能改善后再实施手术治疗。根据国外资料统计,应用体外膜式氧合病例中,
先天性膈疝784例,成活率62%;胎儿持续
肺动脉高压573例,成活率88%。渡过48h后延长至1周内手术者,手术成活率显著高于生后24~48h内急症手术者。但Wilson等将10l例高危
先天性膈疝分为2组,55例即刻手术,46例生后先用体外膜式氧合稳定治疗24~36h再行手术;对比发现,两组的存活率无显著性差异,延迟手术组并用术前体外膜式氧合未能改善存活率,只能减少晚期死亡和肺部后遗症。
总之,虽然外科手术治疗先天性胸
腹裂孔已有70余年的历史,而且近年来在新生儿期病儿的转送、诊断、术前术后处理以及呼吸循环功能障碍的处理、新生儿麻醉和手术技术等方面亦取得了长足进展,使很多的病儿得到了适当的外科治疗,其生存率也显著提高。但是,本病总病死率仍居高不下,特别是出生6h内发病的危重病例病死率非常高,仍是临床医师比较棘手的疾病之一。